RawVegetable’s
Extended Information
Chromatography quality control is a critical step in any
biological mass spectrometry experiment. Several freely available tools tailored
toward shotgun proteomics are available, such as RawMeat (Vast Scientific),
which is probably the most widely adopted, but has been discontinued for some
years. In analogy to RawMeat, we developed RawVegetable, a tool for general
proteomics QC with a focus on XL-MS. RawVegetable includes all RawMeat QC
features plus deals with other standard formats such as *.mzML
and presents several key unique features presented below.
RawVegetable works by loading mass spectrometry data, such as Thermo RAW
files, *.mzML, *.ms1 or Agilent files as input. After
the spectra has been loaded, the chromatographic profile is displayed and all
files loaded are listed on the left side, where the user can choose which ones
to view (Figure 1). Once the data has been loaded, there are a few
analyses the user can make, such as a charged chromatogram, a TopN distribution, and a search for XL artefacts. Loading PatternLab for proteomics [1] *.xic and *.plp or SIM-XL [2] output files enables a reproducibility analysis. All
these features are better described in the following sections.
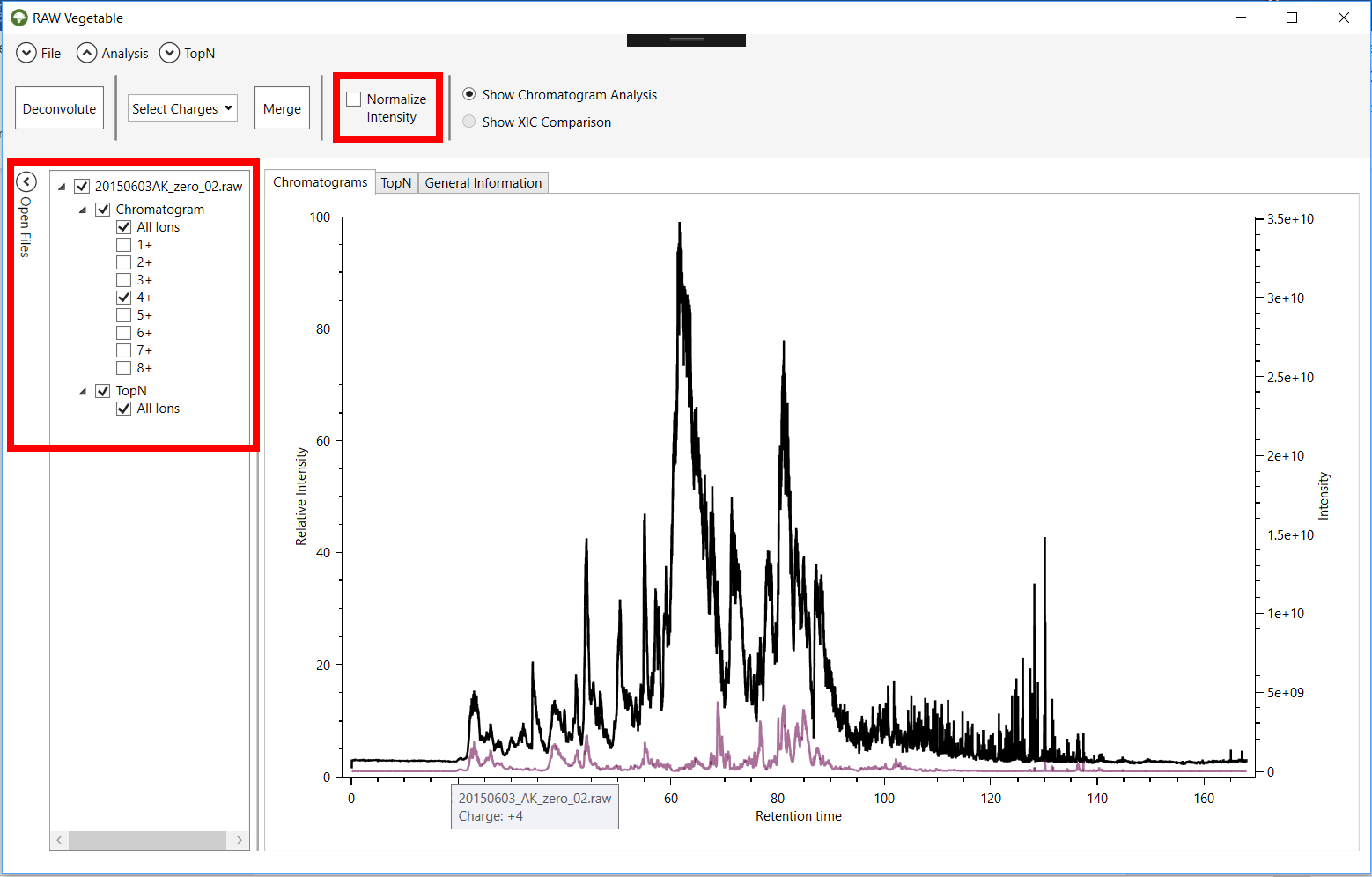
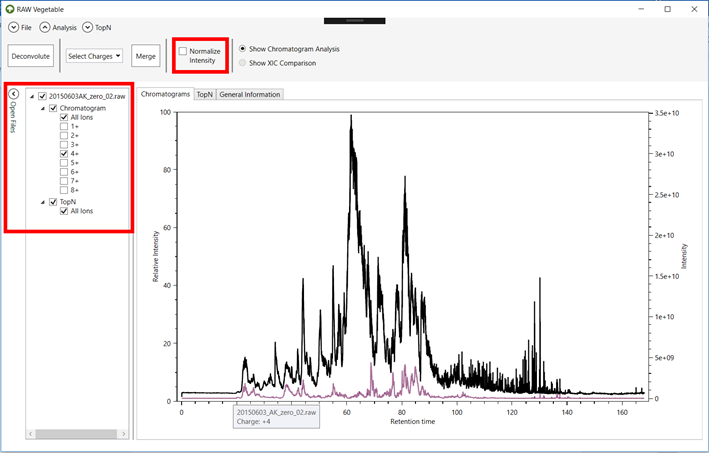
Figure 1 -
RawVegetable’s screen after the spectra file have been loaded. On the left side
it is possible to see the menu where the user can check which files they wish
to view on the screen.
Another analysis the user can make once the spectra files have been
loaded is a preliminary search for contaminants. This is based on a list of
contaminants described by Keller et al [3], which lists around
700 common contaminants found in mass spectrometry experiments, both with ESI
and MALDI ion sources; this list also has a description of the contaminants and
the peaks for which to look in mass spectra. The search RawVegetable performs
takes all MS1 spectra in a file and looks for all the peaks listed as
contaminants for ESI experiments. This can take a few minutes depending on the
number of MS1 spectra in the file and results in a table of contaminants and
the percentages of spectra they were found in (Figure 2). This search can give an idea of possible
contaminants and what to look for in a deeper analysis.
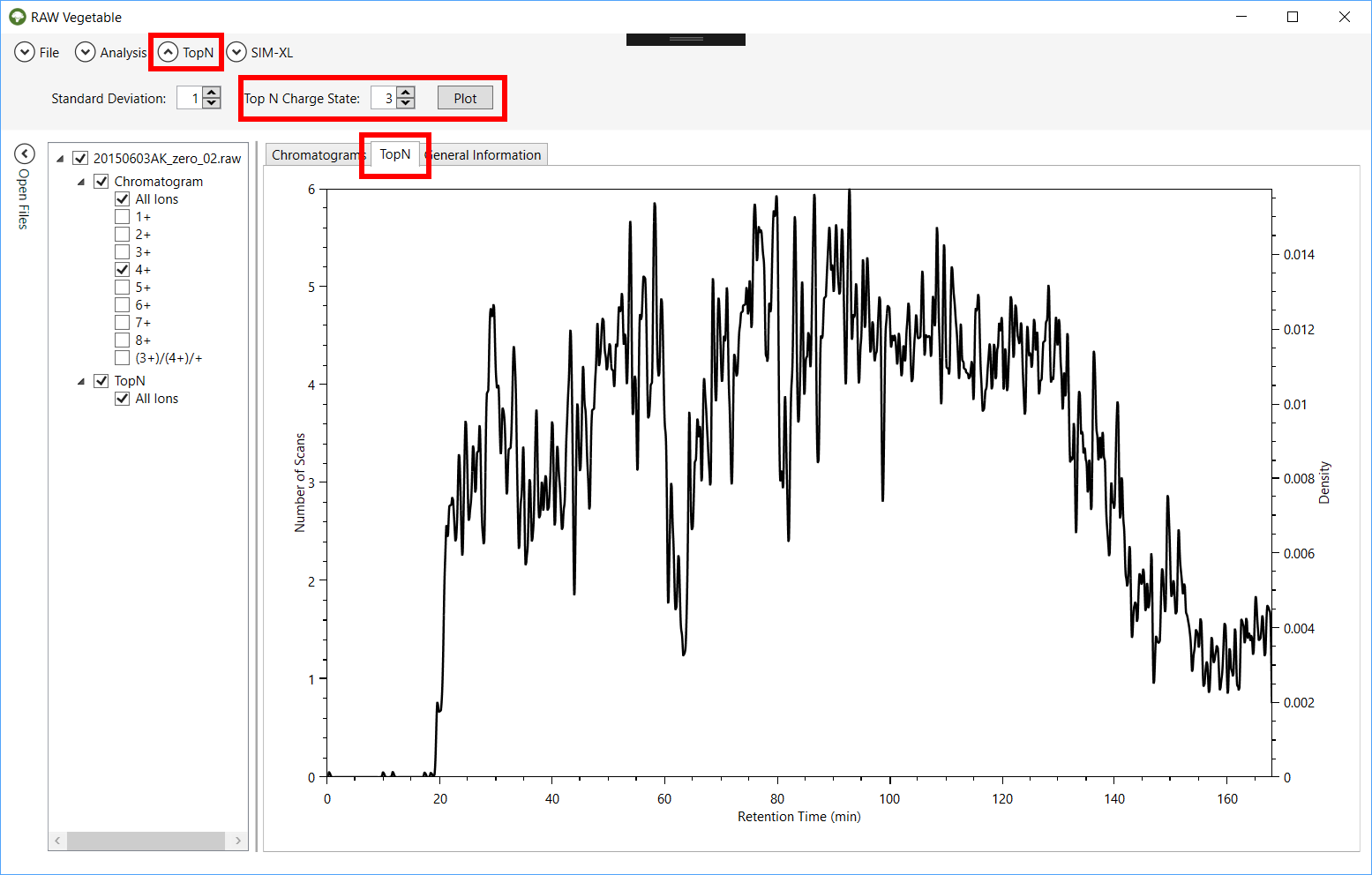
Figure 2 - Resulting table from the
preliminary contaminant search showing possible molecules that had their
respective peaks flagged in more than 50% of the MS1 scans.
Tables with general statistics such as number of MS and MS/MS spectra,
duty cycle average time and chromatography total time and histograms showing
the distribution of precursors’ m/z are also available.
1. Charged Chromatogram
Cross-linked peptides will mostly present higher charge states
than the linear peptides found in traditional proteomic experiments, due to the
existence of a second tryptic peptide in the molecule [7]. Moreover,
cross-linked peptides are typically less abundant than linear ones, making
optimization imperative. As such, chromatographic optimizations with existing
tools are severely limited in the face of XL-MS experiments as their viewers
cannot independently account for highly charged peptide ions.
This charge state chromatogram module is tailored towards
optimizing chromatography for highly charged ion species by independently
plotting the chromatographic profile for each charge state.
In order to do this, all MS1 spectra from each file loaded are
deconvoluted using the YADA algorithm [4]. Deconvolution algorithms consist
of simplifying a spectrum by merging all isotopic and charge envelopes of a
molecule into a single peak.
Isotopic envelopes occur because organic molecules are composed mainly
by carbon, which is present in nature with a mass of 13Da instead of 12Da 1% of
times. Since cross-linked peptides can contain many carbons, the chance of some
of them being 13C is quite high. This ends up creating various peaks
of the same molecule with the mass difference of a neutron between them, as
shown in Figure 3. The fact
that this mass difference is known is what allows isotopic envelopes to be used
for the determination of the charge of the species represented there, using the
following equation:
Here, Peak 1 and Peak 2 are the m/z values of two
consecutive peaks in an isotopic envelope (Figure 3) and the
neutron mass is around 1Da.
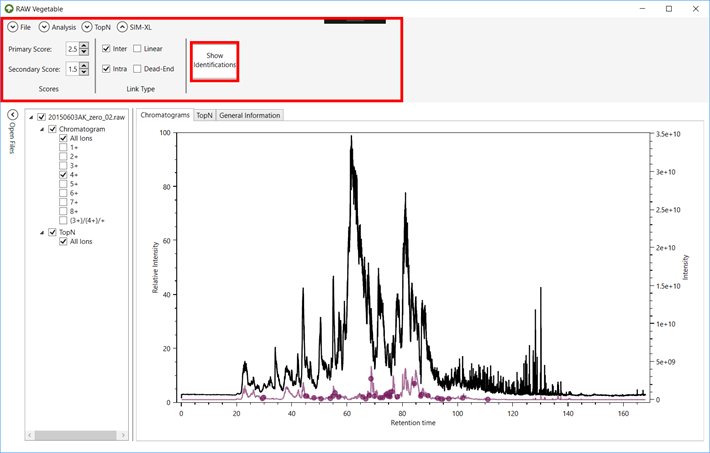
Figure 3 - Representation of an isotopic
envelope of a molecule with a lot of carbon atoms. Using equation (1), we
discover that the charge of this molecule is 1+, so its mass ranges from 700Da
to 712Da, depending on the number of 13C it has. In this case, most
molecules have six 13C, as the most intense peak is the with is the
one that has a difference of around 6Da from the first peak.
This algorithm results in a list of all envelopes found in each
spectrum, which contains their respective charge and the summed intensities of
all peaks in the envelope. With this information, it is possible to choose a
single charge and build a chromatogram using the intensities of all envelopes
with the selected charge, as shown in Figure 4. This new chromatogram
can give insights into when most cross-linked peptides are eluting, as they
usually appear with charges 3+ and 4+. The chromatography gradient can thus be
altered to prioritize such regions, enrich populations of desired charge
states, which could help in the acquisition of more MS2 spectrum from
cross-linked species in lieu of conventional, linear peptides.
In the GUI, the user can then choose which charge chromatogram to
view on the left side of the screen, by checking the boxes of each file, as in Figure 4. It is also possible to normalize the chromatograms on the screen
in order to see them all as relative intensities and merge charged chromatograms
to see them as one, as in Figure 4.

Figure 4 - RawVegetable's screen after
the deconvolution algorithm has run. Represented in black is the full
chromatogram of the file selected in the menu on the left; in purple is the
chromatogram considering only species with charge 3+. On the top menu it is
possible to select charges to merge, as well as normalize the intensities of
the chromatograms.
If the search for cross-links has already been performed, it is
possible to identify the regions where most of them appeared by loading the
SIM-XL output file into RawVegetable. This will result in all identifications
being represented in the chromatograms as small circles at the retention time
where the scan was extracted. The user can filter the identification by link
type and by the scores calculated by SIM-XL (Figure 5).
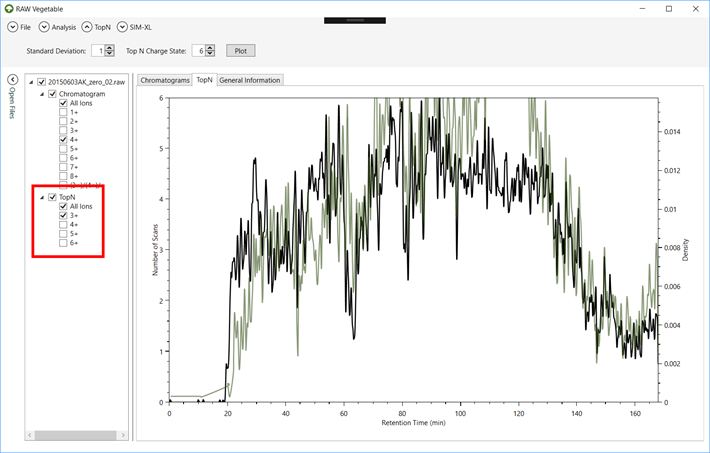
Figure 5 - The software's screen after a
SIM-XL output file has been loaded. Here the full chromatogram is represented
in black and the charge 4+ is in purple. The purple dots indicate when a
cross-link identified with charge 4+ eluted. It is possible to change the score
and link type filtering in the top menu.
2. TopN Distribution
This module shows the density estimation of MS2 scans per duty
cycle along the chromatography. This allows pin-pointing retention time
intervals that could lead to over- or under-sampling, so the necessary gradient
adjustments can be done.
In order to do this, the algorithm applies Kernel Density
Estimation (KDE) using a Gaussian distribution as the kernel function.
Essentially what the algorithm will do is assign a Gaussian for each duty
cycle, which will then be multiplied by the number of MS2 scans in that cycle.
This will result in several Gaussians overlapping, which will then be summed,
generating a density estimation plot, as in Figure 6. This way, every
gaussian influences all nearby points in the resulting plot, even if minimally.
Given a dataset composed of the retention time of all MS1 scans,
that is, the initial retention time of all duty cycles, as the values of x in
a plot, the KDE function used to find their respective y value to
generate the density estimation plot is the following [5,6]:
Where n is the number of duty cycles
(which is the same as the number of MS1 scans); h is a smoothing factor
called bandwidth; ![]() is the kernel function, where x is the
retention time of the current point in the KDE being calculated and xi
is the x values of the rest of the dataset, that is, the retention time
of other duty cycles. The kernel function with x-xi as
parameter represents the influence of all gaussians in the determination of
that specific point being calculated, as it returns the intensities of
overlapping regions (Figure 6) to be
summed, as represented by the summation (∑) which goes from the first
retention time (i = 1) to the
last (i = n).
is the kernel function, where x is the
retention time of the current point in the KDE being calculated and xi
is the x values of the rest of the dataset, that is, the retention time
of other duty cycles. The kernel function with x-xi as
parameter represents the influence of all gaussians in the determination of
that specific point being calculated, as it returns the intensities of
overlapping regions (Figure 6) to be
summed, as represented by the summation (∑) which goes from the first
retention time (i = 1) to the
last (i = n).
Figure 6 - Graphical representation of
how the Kernel Density Estimation works. For each duty cycle (DC1, DC2 and DC3)
a standard normal distribution is built and then multiplied by its respective
number of MS2 scans, generating all these different gaussian with overlapping
regions, which will then be summed to create a density estimation plot, such as
the represented in orange.
The bandwidth (h) is a smoothing factor
which greatly influences the resulting plot and should be as small as possible
without causing the plot to be oversmoothed. It can be a tricky parameter to
calculate, but for a Gaussian kernel the following equation can be used [7]:
Where σ is the standard deviation of the
distribution, which is user-defined, but has a default value of 1; n is
the total number of duty cycles.
The kernel function (K(x)) used is a
normal (Gaussian) distribution with a mean value of 0, which is defined by the
following general form [8]:
Where x is ![]() as defined by the KDE equation and σ2
is the variance, which is the square of the standard deviation, and has the
same value as in the h formula.
as defined by the KDE equation and σ2
is the variance, which is the square of the standard deviation, and has the
same value as in the h formula.
The KDE function can then be represented by
the following equation:

Applying this function for all duty cycle
retention times results in a plot such as the one shown in Figure 7. This plot permits optimizing the
chromatography gradient so that the TopN (MS2 scans
in a duty cycle) distribution is as wide and homogeneous as possible through
time. This optimization can also help shortening the extremities of the
chromatography run, thus shortening the time of the experiment.

Figure 7 - RawVegetable's screen after
the TopN distribution has been calculated, with the
user being able to choose the standard deviation to be used during the
calculation. In this case the maximum number of scans in a duty cycle was six
spectra and that the equipment did not maintain efficiency throughout the run.
It is also possible to calculate a KDE using
only MS2 scans with precursors of a specific charge
(Figure 8), to have a
better notion of where heavily charged species elute more frequently, similar
to the charged chromatograms.

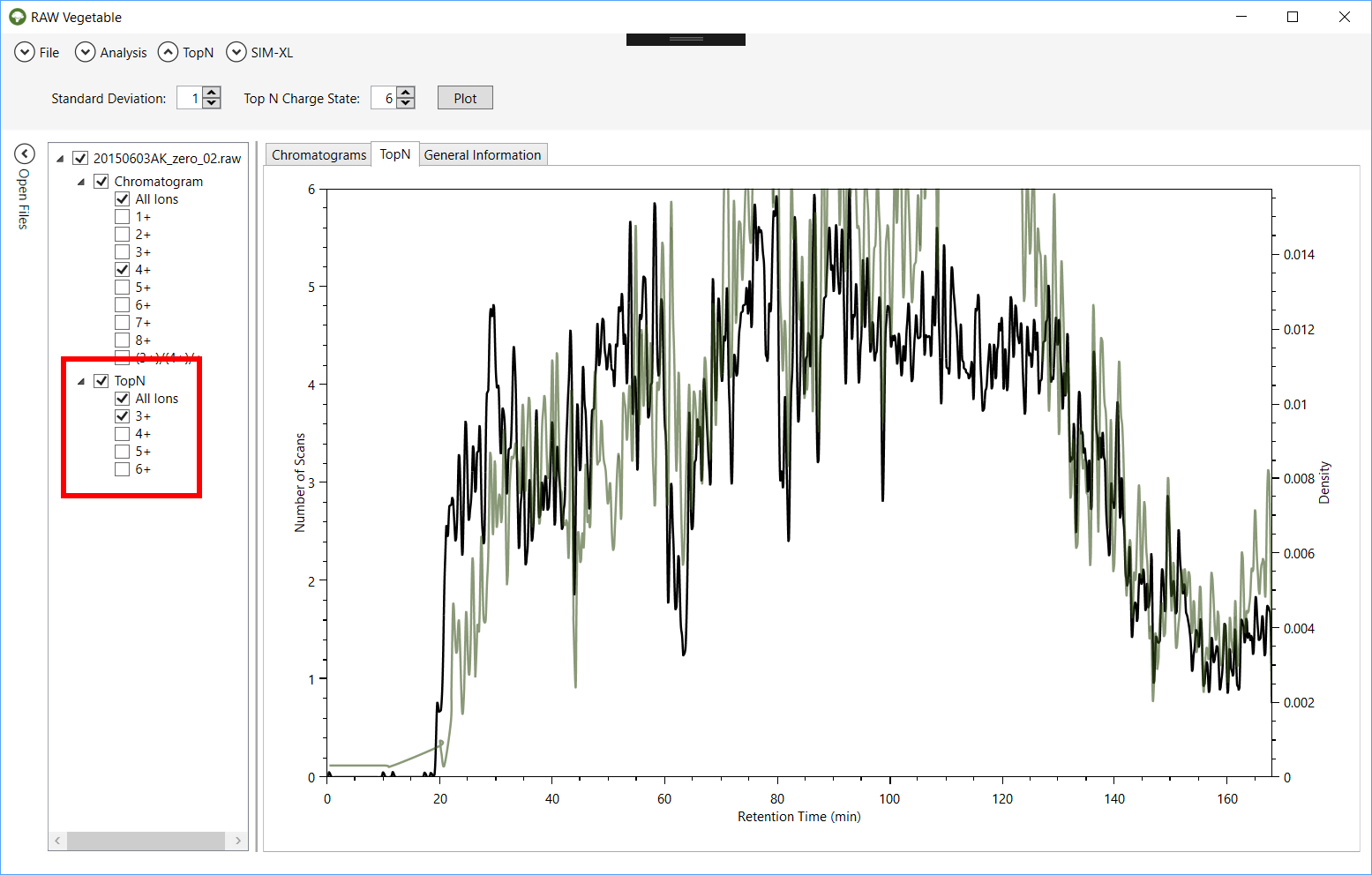
Figure 8 - TopN
distribution with specific charge state selection. It is possible to view the TopN distribution of species with a single selected charge,
as shown by the green curve, which represents the distribution for charge 3+
precursors; in black is the total TopN distribution.
For experiments where the TopN
number has not been set and instead only the time for the duty cycle has been
specified in the equipment, it is interesting to look at the performance of the
run by viewing the TopN distribution as a frequency plot (Figure 9 (A)). Zooming in regions of interest shows
exactly how many scans were obtained in specific duty cycles (Figure 9 (B)). It is also
possible to compare the density estimation with the frequency plot by checking
both on the menu on the left side of the screen.

Figure 9 - (A) Frequency plot of the MS2
scans during the experiment. (B) Zooming in shows that this plot computes
absolute values for the number of scans instead of a distribution as was the
case before.
Another application becomes handy especially when using very
high-scan rate instruments such as a Thermo Lumos or HFX. The user might set a TopN and get a very homogeneous distribution throughout the
run. This could mean that the equipment is being very efficient and even
slightly saturated, meaning that the acquisition method could be modified to
acquire more MS2 scans per duty cycle, and so take full advantage of the
equipment.
3. XL-Artefact
Recently, Giese et al. [9] described the presence of some high
scoring identifications wrongly taken for interlink peptides owing to the
non-covalent dimerization of a linear peptide and an intralinked
one during the ionization step of the mass spectrometry, when the molecules are
going to the gas phase. Based on this, we added a feature which
flags possible species such as these, which we called XL-Artefacts.
RawVegetable
uses a SIM-XL output file to identify whether an artefact is at hand by
verifying if XICs of all possible separate species (linear α-peptide and intralinked β-peptide and the inverse) are found
during the same retention time range with charges 2+ and 3+. A score is then
assigned to the possibly wrong inter-linked XL. This score is calculated as
follows: if a linear α-peptide and an intralinked
β-peptide or an intralinked α-peptide and a
linear β-peptide of any charges have their XICs extracted successfully,
one point plus a bonus based on the relative intensities of the XICs are summed
to the score. If all four species have valid XICs, then an extra bonus is
given. Based on this scoring system, cross-links with scores higher than 1
should already be looked at more closely, however tests made with proteins of
known crystallographic structure showed that species
with scores higher than 2.5 are the ones more prone to be artefacts.
The result
of this algorithm is a table with all interlinked peptides found, with their
respective XL-Artefact score and SIM-XL scores listed. The user can also view
these possible artefacts represented in the charged chromatograms as red dots,
as shown in Figure 10.

Figure 10 - A charge 3+ (in green) and charge
4+ (in blue) chromatograms. The green and blue dots indicate where cross-linked
species were identified with charges 3+ and 4+, respectively; the red dots
indicate possible XL-artefacts that should be closely analysed.
We exemplify the usefulness of our artefact score on the XL
K499-S709 for the HSP90 protein as per one of our experiments. In our example,
the XL XIC curve is shown in yellow (Figure 11). The
crosslink information is not in agreement with an open conformation model of
the HSP90; the Euclidean distance between K499-S709 is of 26Å, while the
maximum distance for the DSS crosslinker is of 17Å. The error becomes even
higher when comparing this crosslink’s distance constraint result with the
closed conformations models of HSP90, which goes up to 56Å. These results
corroborate with a wrong identification, due to an artefact of non-covalent
association in gas phase. Our
“XL-Artefact Analysis” flags this event as it detects the XICs of other
possible species. Figure 12 shows in brown and pink the curves for the XICs of the alpha
sequence as linear peptide with charges 3+ and 2+; in lilac and green the
curves for the XICs of the beta sequence as an intralinked peptide with charges 3+ and 2+; and in
yellow, the curve for the XIC of the identified interlink. The results
demonstrate that all these peptides eluted during the same time as the
“identified interlink”; we also note that the separate species present
significantly higher intensities, which strongly suggests that the interlink is
in fact a linear alpha peptide non-covalently bound to the intralinked
beta peptide in the gas phase.

Figure 11 - XIC Curve for cross-link K499-S709 of the HSP90 protein.

Figure 12 - XIC curves for the individual peptides
of the cross-link K499-S709 eluting at the same
time range as the identified interlink. In brown and pink, the curves for the
XICs of the alpha sequence as linear peptide with charges 3+ and 2+; in lilac
and green the curves for the XICs of the beta sequence as an intralinked
peptide with charges 3+ and 2+; and in yellow, the curve for the XIC of the
identified interlink.
4. Reproducibility Analysis
This module performs all pairwise comparisons of XICs between
identifications of different runs, which allows a general view of the
chromatographic reproducibility for all runs and therefore to quickly locate
problematic MS run.
In order to perform this analysis, RawVegetable
needs the PatternLab’s *.xic
or *.plp result, or the SIM-XL output files as input.
The software will then group all common peptides between the files and proceed
to compare their XICs (which should already have been calculated and stored in
the input files). This comparison between common peptides is made for each
pairwise combination of files, with the XICs of one file representing the x
values and the XICs of the other file, the y values. With these values
in hand, it is possible to calculate a similarity score between the files; the
user can choose the following metrics to use for the score: r2, k
or k2.
The r2 metric is the coefficient of
determination and calculates how well a set of points fits a linear regression;
in this case, a line represented by the equation y = x. The general
formula for r2 is the following [10]:

Where yi
the actual value of y being used; ŷi
is the predicted value of yi,
following the equation y = x and ӯ is the mean of the y
values. In this case, the closer the r2 is to 1, the more
similar are files being compared.
The k metric is the Euclidean distance between the XIC
values of each file. The XIC values of the first file will be represented by
the vector x, where x = (x1, x2, …, xn) and the second file by y¸ where y
= (y1, y2, …, yn).
First the values are normalized to be within a range of 0 to 1 and then the
Euclidean distance can be calculated using the following equation [11]:

Using this metric gives an information which
is the inverse of r2; the closer the k value is to 0,
the more similar are the files.
Another metric that can be used is the square
of the k value (k2), which is calculated the same way,
but accentuates the similarities between files.
The calculation of these scores produces a
heatmap comparing files using the chosen metric. The plots generated using the
different score systems are shown in
Figure 13.
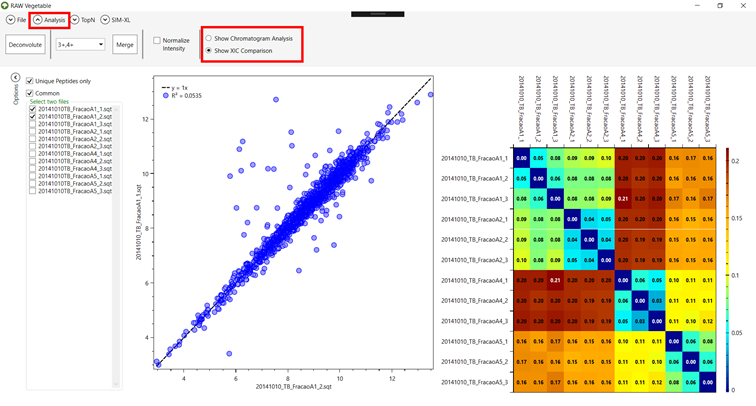
It is also possible compare two specific files
by marking them on the left side of the screen. This will result in a dot plot
of all common peptides between the files according to their XIC values, along
with a trend line and the score value, as in Figure 14.
This analysis allows to easily pinpoint
problematic experiments and determine whether a replicate should be used during
the research or not.



Figure 13 - The heatmaps generated using k (A), k2
(B) and r2
(C) as score metrics. It is possible to see that while the colour system
changes, the information given is the same, the technical replicates (in groups
of three in the order shown) are very similar, while some of the biological
replicates are quite different.

Figure 14 - Dot plot comparing the XIC of
common peptides of two different files, with the score shown in the left as the
metric k,
and a trendline for the equation y
= x.
5. Precursor ion dissociation efficiency
In this feature, the proportion between the intensity of the
precursor peak in a MS2 scan and the total intensity of all peaks in the same
scan is calculated. The motivation is to assess how much of the MS2 scan is
composed of the precursor and thus evaluate the collisional energy and
dissociation process efficiency. In brief, the software does this process for
every MS2 scan to ultimately present the results as histogram (Figure 15). The higher
the distribution’s density is found near zero, the more efficient the
fragmentation is, as the precursor peak is responsible for less of the total
ion current of the scan.

Figure 15 - Histogram of the ratios between precursor intensity and total ion current of a MS2 scan, which can give some information on the fragmentation efficiency of an acquisition. The different colours represent different experiments being compared in the same plot.
References:
[1] P.C. Carvalho, D.B. Lima, F.V. Leprevost, M.D.M. Santos, J.S.G. Fischer, P.F. Aquino, J.J.
Moresco, J.R. Yates, V.C. Barbosa, Integrated
analysis of shotgun proteomic data with PatternLab
for proteomics 4.0, Nat. Protoc. 11 (2015) 102–117.
https://doi.org/10.1038/nprot.2015.133.
[2] D.B. Lima, J.T.
Melchior, J. Morris, V.C. Barbosa, J. Chamot-Rooke,
M. Fioramonte, T.A.C.B. Souza, J.S.G. Fischer, F.C. Gozzo, P.C. Carvalho, W.S. Davidson, Characterization of
homodimer interfaces with cross-linking mass spectrometry and isotopically labeled proteins, Nat. Protoc. 13
(2018) 431–458. https://doi.org/10.1038/nprot.2017.113.
[3] B.O. Keller, J. Sui,
A.B. Young, R.M. Whittal, Interferences and
contaminants encountered in modern mass spectrometry, Anal. Chim.
Acta. 627 (2008) 71–81. https://doi.org/10.1016/j.aca.2008.04.043.
[4] P.C. Carvalho, T. Xu,
X. Han, D. Cociorva, V.C. Barbosa, J.R. Yates, YADA:
a tool for taking the most out of high-resolution spectra, Bioinformatics. 25
(2009) 2734–2736. https://doi.org/10.1093/bioinformatics/btp489.
[5] M. Rosenblatt, Remarks
on Some Nonparametric Estimates of a Density Function, Ann. Math. Stat. 27
(1956) 832–837. https://doi.org/10.1214/aoms/1177728190.
[6] E. Parzen,
On Estimation of a Probability Density Function and Mode, Ann. Math. Stat. 33
(1962) 1065–1076. https://doi.org/10.1214/aoms/1177704472.
[7] B.W. Silverman,
Density estimation for statistics and data analysis, Chapman & Hall/CRC,
Boca Raton, 1998.
[8] J.K. Patel, C.B. Read,
Handbook of the normal distribution, 2nd ed., rev.expanded,
Marcel Dekker, New York, 1996.
[9] S.H. Giese, A. Belsom, L. Sinn, L. Fischer, J. Rappsilber,
Noncovalently Associated Peptides Observed during Liquid Chromatography-Mass
Spectrometry and Their Effect on Cross-Link Analyses, Anal. Chem. 91 (2019)
2678–2685. https://doi.org/10.1021/acs.analchem.8b04037.
[10] T.O. Kvalseth, Cautionary Note about R 2, Am. Stat. 39 (1985)
279. https://doi.org/10.2307/2683704.
[11] H. Anton, Elementary
linear algebra, 7th ed, John Wiley, New York, 1994.